
Complement factor H levels in steady state sickle cell anaemia
Keywords:
Complement factor H, sickle cell anaemia, alternative pathwayAbstract
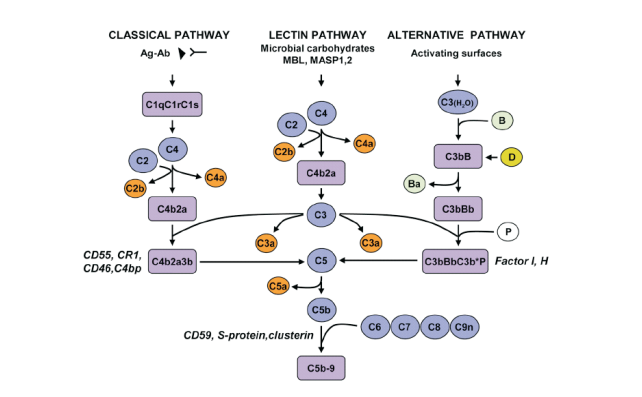
Objective: The red cell membrane of sickle cell anaemia is vulnerable to attack from the alternative complement pathway. The activation of the alternative complement pathway is initiated by externalization of phosphatidylserine on red cell membrane. Serum and cell bound regulators normally prevent amplification of the cascade. However, red blood cells in sickle cell anaemia appear to be exposed and the cell lysing membrane attack complex is ubiquitous on irreversible sickle red blood cells. It is possible that there are deficiencies (either functional or quantitative) of complement regulators. In this study the quantitative defects of the most abundant serum phase regulator, complement factor H in sickle cell anaemia was investigated.
Methods: We compared the plasma levels of complement factor H (a serum phase regulator of the alternative pathway) in 61 steady state Hb SS with 60 healthy Hb AA using an enzyme linked immunosorbent assay to analyze complement factor H level in the plasma. The full blood count parameters were estimated using flow cytometry.
Results: There was no significant difference in the serum complement factor H levels between the steady state Hb SS and healthy Hb AA. Significant inverse relationships existed between complement factor H, total white cell count, granulocyte cell count and platelet count as well as significant direct relationships between complement factor H, haematocrit, and the haemoglobin concentration.
Conclusion: Complement factor H in patients with sickle cell anaemia who are in steady state is not significantly lower than in controls.
References
Brousse V, Makani J, Rees DC. Management of sickle cell disease in the community. BMJ. 2004; 348: 1765.
Ballas SK, Kesen MR, Goldberg MF, et al. Beyond the Definitions of the Phenotypic Complications of Sickle Cell Disease: An Update on Management. The Scientific World Journal. 2012; 2012:949535. doi:10.1100/2012/949535
Steinberg MH. Sickle cell anemia, the first molecular disease: overview of molecular etiology, pathophysiology, and therapeutic approaches. The Scientific World Journal. 2008; 8:1295–1324. DOI:10.1100/tsw.2008.157
Eaton, W.A. and Hofrichter, J. Sickle cell hemoglobin polymerization. Adv. Protein Chem.1998; 40, 63–280.
Lei H, Karniadakis GE. Predicting the morphology of sickle red blood cells using coarse-grained models of intracellular aligned hemoglobin polymers. Soft matter. 2012;8(16):10.1039/C2SM07294G. doi:10.1039/C2SM07294G.
De Jong, K., Larkin, S.K., Styles, L.A., Bookchin, R.M., and Kuypers, F.A.
Characterization of the phosphatidylserineexposing subpopulation of sickle cells. Blood. 2001; 98, 860–867.
Lubin, B., Kuypers, F., and Chiu, D.Lipid alterations and cellular properties of sickle red cells. Ann. N.Y. Acad. Sci.1989; 565, 86–95.
Wang RH, Phillips G, Medof ME, Mold C. Activation of the alternative complement p a t h w a y b y e x p o s u r e o f
p h o s p h a t i d y l e t h a n o l a m i n e a n d phosphatidylserine on erythrocytes from sickle cell disease patients. Journal of Clinical Investigation. 1993;92(3):1326-1335.
Test S,Woolworth V. Defective regulation of complement by the sickle erythrocyte: evidence for a defect in control of membrane attack complex formation. Blood, 1994. 83(3), 842-852.
Meulenbroek EM, de Haas M, Brouwer C, Folman C, Zeerleder SS, Wouters D. Complement deposition in autoimmune hemolytic anemia is a footprint for difficult-todetect IgM autoantibodies. Haematologica.
0 1 5 ; 1 0 0 ( 1 1 ) : 1 4 0 7 - 1 4 1 4 . doi:10.3324/haematol.2015.128991.
DeZern AE, Brodsky RA. Paroxysmal Nocturnal Hemoglobinuria: A Complement-Mediated Hemolytic Anemia. Hematology/oncology clinics of North America. 2015;29(3):479-494. doi: 10.1016/j.hoc.2015.01.005.
Walport MJ. Complement. N Engl J Med. 2001; 344: 1140–1144.
Zewde N, Gorham RD, Jr., Dorado A, Morikis D. Quantitative Modeling of the Alternative Pathway of the Complement System. PLoS ONE .2016;11(3): e0152337.
Lubbers R, van Essen MF, van Kooten C, Trouw LA. Production of complement components by cells of the immune system. Clinical and
Experimental Immunology. 2017;188(2):183194. doi:10.1111/cei.12952.
Morgan HP, Schmidt CQ, Guariento M, et al. Structural basis for engagement by complement factor H of C3b on a self-surface. Nature structural & molecular biology. 2011;18(4):463470. doi:10.1038/nsmb.2018.
Pangburn M. Cutting Edge: Localization of the Host Recognition Functions of Complement Factor H at the Carboxyl-terminal. Implications of Hemolytic Uremic Syndrom. J I m m u n o l . 2 0 0 2 ; 1 6 9 : 4 7 0 2 - 4 7 0 6 d o i : https://doi.org/10.4049/jimmunol.169.9.4702
Leitao MF, Vilela MM, Rutz R, Grumach AS, Condino-Net A, Kirschfink M. complement factor I deficiency in a family with recurrent infections. Immunopharmacology.1997;38(12):207-213 https://doi.org/10.1016/S0162-
(97)00080-5
Wilson WA, Hughes GR, Lachmann PJ.
Deficiency of factor B of the complement system in sickle cell anaemia. British Medical Journal. 2003;1(6006):367-369.
Quirolo K, Vichinsky E. Haemoglobin disorders. In: Behrmen RE, Kliegman EM, Jenson HB
(eds). Nelson Text Book of Paediatrics. 17th ed.
Philadephia. Saunders Company 2004; 1623-
Atkinson JP, Goodship THJ. Complement factor
H and the haemolytic uremic syndrome.
J E M . 2 0 0 7 ; 2 0 4 ( 6 ) : 1 2 4 5 - 1 2 4 8 d o i .
1084/JEM20070664
Kavanagh D, Goodship TH, Richards A. Atypical Hemolytic Uremic Syndrome. Seminars in Nephrology. 2013;33(6):508-530. doi:
1016/j.semnephrol.2013.08.003.
Miao D, Li D-Y, Chen M, Zhao M-H. Platelets are activated in ANCA-associated vasculitis via thrombin-PARs pathway and can activate the alternative complement pathway. Arthritis Research & Therapy. 2017; 19:252. doi:10.1186/s13075-017-1458-y.
Downloads
Published
How to Cite
Issue
Section
License
Copyright (c) 2023 Research Journal of Health Sciences

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Research Journal of Health Sciences journal is a peer reviewed, Open Access journal. The Journal subscribed to terms and conditions of Open Access publication. Articles are distributed under the terms of Creative Commons License (CC BY-NC-ND 4.0). (http://creativecommons.org/licences/by-nc-nd/4.0). All articles are made freely accessible for everyone to read, download, copy and distribute as long as appropriate credit is given and the new creations are licensed under the identical terms.
